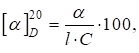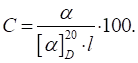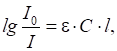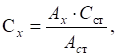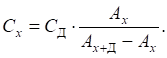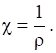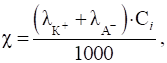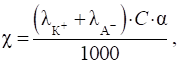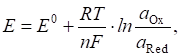|
|
|
АНАЛІТИЧНА ХІМІЯ Електронний посібник |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
РОЗДІЛ 4. ОСНОВИ ФІЗИКО-ХІМІЧНИХ
МЕТОДІВ АНАЛІЗУ |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
4.1. Загальна характеристика фізико-хімічних методів аналізу 4.1.1. Поняття про
фізико-хімічні методи аналізу 4.1.2. Класифікація
фізико-хімічних методів аналізу 4.2.1.1. Поняття про
рефрактометрію 4.2.1.3.
Рефрактометр і правила користування ним 4.2.1.4.
Рефрактометричне визначення складу двокомпонентних систем 4.2.2.1. Поняття про
оптичну активність 4.2.2.2. Розрахунок
величини питомого обертання 4.2.3. Фотоелектроколориметрія 4.2.3.1. Закон Бугера-Ламберта-Бера 4.2.3.2. Фотоелектроколориметр 4.2.3.3. Методи
визначення концентрації розчинів за допомогою фотоелектроколориметрії 4.2.4.1. Теоретичні
основи спектрофотометрії 4.3. Електрохімічні методи аналізу 4.3.1.1. Теоретичні
основи кондуктометрії 4.3.1.4. Кондуктометричне титрування 4.3.2.1. Теоретичні
основи потенціометрії 4.3.2.2. Електроди
для потенціометрії 4.3.2.4.
Потенціометричне титрування 4.4. Хроматографічні методи
аналізу 4.4.1. Сутність та класифікація хроматографічних методів аналізу 4.4.1.1. Теоретичні
основи хроматографічного аналізу 4.4.1.2.
Класифікація хроматографічних методів аналізу 4.4.2. Основні методи
хроматографічного аналізу 4.4.2.1. Паперова тонкошарова хроматографія 4.4.2.3. Рідинна
хроматографія 4.4.2.4. Йоннообмінна хроматографія 4.1. Загальна характеристика
фізико-хімічних методів аналізу 4.1.1. Поняття про фізико-хімічні методи
аналізу
Сучасна
промисловість: хімічна, металургійна, фармацевтична, харчова вимагає високої чутливості методів аналізу,
їх селективності та експресивності виконання. Ще однією важливою вимогою
сучасності є управління технологічними
процесами в автоматичному режимі й дистанційно. Ці завдання певною мірою
вдається вирішувати за допомогою фізико-хімічних методів аналізу. Сучасні
фізико-хімічні методи аналізу дозволяють виконувати водночас якісний і
кількісний аналіз і застосовувати комп’ютерну техніку для обробки
результатів. Поширення
фізико-хімічних методів аналізу, в першу чергу, пов’язано з тим, що ці методи
мають значно більшу чутливість
порівняно з хімічними методами. Висока чутливість фізико-хімічних методів аналізу
призводить до їх широкого використання у виробництві речовин високої і
надвисокої чистоти, абсолютно необхідних сучасній науці і техніці, вони
можуть забезпечити достатньо надійне визначення домішок з вмістом 10-8–10-10%.
Якщо звичайними хімічними методами можна визначити концентрацію речовини 10-5
моль/см3, то для деяких фізико-хімічних методів визначуваний
мінімум менший приблизно на 5 порядків, тобто 10-9 – 10-10 моль/см3. Основні переваги фізико-хімічних методів аналізу: ► висока
чутливість; ►
селективність; ►
об’єктивність; ► висока
швидкість (експресність); ► можливість
комп’ютеризації й автоматизації процесу. Однак
фізико-хімічні методи аналізу мають і недолік – високу ціну приладів, що їх
застосовують. 4.1.2. Класифікація фізико-хімічних
методів аналізу В основу
класифікації фізико-хімічних методів аналізу покладено фізичні властивості
речовини та їх природу й величину. Фізико-хімічні методи аналізу поділяють на
такі групи:
До таких
параметрів належать: інтенсивність випромінювання збуджених атомів,
поглинання монохроматичного випромінювання, вимірювання показника заломлення
світла, кута обертання площини поляризованого світла ті інші.
Контрольні питання 1. Які методи аналізу називають фізико-хімічними? 2. Які переваги фізико-хімічних методів аналізу? 3. На які групи поділяють фізико-хімічні методи аналізу? 4. Які методи аналізу називають оптичними? 5. Які методи аналізу належать до електрохімічних? 6. Що таке хроматографічні методи аналізу? 4.2.1. Рефрактометрія 4.2.1.1. Поняття про рефрактометрію
Рефрактометричний
аналіз дає можливість визначати одну з найважливіших характеристик речовини –
показник заломлення. Показник заломлення дозволяє ідентифікувати речовину,
розраховувати її молекулярну рефракцію, а потім зробити припущення про будову
речовини. Рефрактометрія ‒ це швидкий, дешевий і надійний метод пізнання чистоти речовини,
саме тому його широко використовують у хімії, біоаналізі
та харчових технологіях. Але оскільки
існують різні речовини з однаковим показником заломлення, необхідно знати,
яка саме аналізується. Наприклад, відомо, що циклогексан і деякі розчини
цукру мають однаковий показник заломлення за температури 20 °С. З іншого боку,
показник заломлення значно залежить від температури, як зазначено вище, на
додаток до тиску та концентрації розчину. Всі ці параметри слід ретельно
контролювати, коли потрібні високоточні вимірювання.
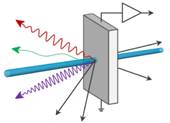
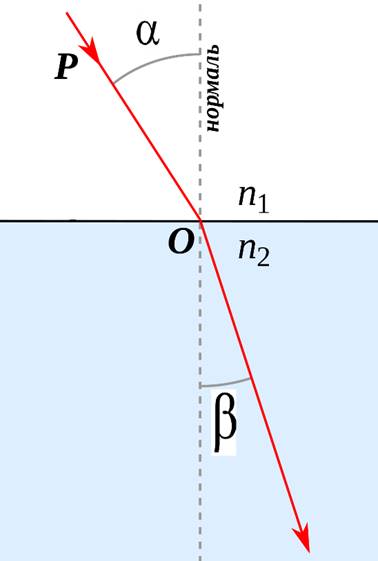
Рис. 4.1. Заломлення кута світла на межі
двох середовищ
На практиці
показник заломлення визначають за довжини хвилі, що відповідає довжині
жовтого випромінювання Натрію (D – лінія, λ = 589 нм) за 20 0С.
Визначаючи показник заломлення, обов’язково вказують температуру й довжину
хвилі 4.2.1.3. Рефрактометр і правила
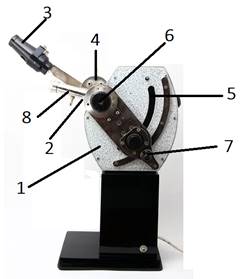
користування ним Показники
заломлення визначають за допомогою рефрактометрів.
Правила користування рефрактометром. Призми рефрактометра 4 протирають ватним тампоном з
ефіром або спиртом. На поверхню нижньої призми скляною паличкою з оплавленим
кінцем наносять 2- 3 краплі досліджуваної рідини й накривають
верхньою призмою. Джерело світла 3 спрямовують так, щоб світло падало у
віконце 2 і рівномірно освітлювало все поле зору. Обертаючи маховик 7,
знаходять в окулярі межу світла й тіні. Якщо ця межа розмита й нечітка, то її
вирівнюють за допомого гвинта 6. За допомогою маховика 7 поєднують межу
світла й тіні з перехрестям сітки та визначають показник заломлення за шкалою
(рис. 4.3). Для юстирування рефрактометра застосовують дистильовану воду, для
якої
Після роботи
рефрактометр слід протерти спиртом, фільтрувальним папірцем. 4.2.1.4. Рефрактометричне визначення складу
двокомпонентної системи Рефрактометричний
аналіз дозволяє встановити склад двокомпонентної системи (наприклад, розчину)
за результатами вимірювання показника заломлення для багатьох стандартних
систем, уміст компонента в яких точно відомий. Так, для визначення в розчині
масової частки розчиненої речовини вимірюють показники заломлення серії
розчинів з відомою масовою часткою розчиненої речовини. Для багатьох розчинів
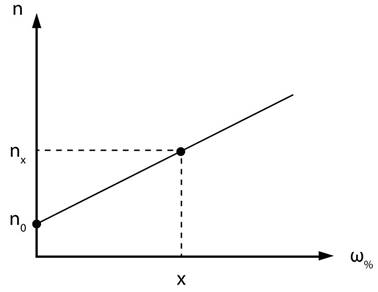
ця залежність прямолінійна. На підставі
отриманих даних будують графік залежності показника заломлення від
концентрації речовини. Далі вимірюють показник заломлення для розчину з
невідомою концентрацією. Для визначення концентрації цього розчину проводять
перпендикуляр від значення показника заломлення до графіка, а від точки
перетину перпендикуляра з графіком проводять перпендикуляр на вісь
концентрацій. Те значення, яке покаже перпендикуляр на осі абсцис, і буде
невідома концентрація (рис. 4.4).
Контрольні питання 2. Що називається показником заломлення? 3. За допомогою яких приладів визначають показник заломлення? 4. Як за допомогою рефрактометричного методу аналізу визначити склад
двокомпонентних систем? 5. Наведіть приклади
застосування рефрактометричного аналізу для харчових продуктів і сировини.
4.2.2. Поляриметрія 4.2.2.1. Поняття про оптичну активність
Оптично активними
є більшість вуглеводів, ефірні олії, оксикислоти. Згідно з
електромагнітною теорією будь-яке джерело світла випускає неполяризоване
випромінювання. Деякі кристалічні речовини, наприклад іспанський шпат, мають
властивість пропускати промені тільки певного напрямку коливань. Унаслідок
проходження світла через такий кристал коливання променів відбувається лише в
одній площині. Із цього матеріалу виготовляють призми Ніколя. Як аналізатор
використовують матеріал турмалін. Різні речовини
по-різному здатні обертати площину поляризації світла. Для порівняльної
оцінки цієї здатності обчислюють величину питомого обертання [α]. Питоме
обертання є константою для кожної оптично активної речовини. Його
розраховують як кут повороту площини поляризації монохроматичного світла, що
проходить відстань 1 дм у середовищі з оптично
активною речовиною за умовної концентрації 1 г/см3. На
величину питомого обертання впливає природа речовини, довжина хвилі
поляризованого світла й температура. Тому величину питомого обертання
відносять до 20 °C і жовтої монохроматичної лінії D спектра Натрію. Величину
питомого обертання позначають 4.2.2.2. Розрахунок величини питомого
обертання Для розрахунку величини питомого обертання речовини, що міститься в розчині, користуються формулою:
де α –
виміряний кут обертання в градусах; L– товщина шару, дм; С – концентрація розчину в грамах речовини на 100 г розчину. Якщо відоме
значення
4.2.2.3. Поляриметр Для
поляриметричного способу визначення концентрації оптично активних речовин
користуються поляриметрами (рис. 4.5). Принципова схема поляриметра базується на тому, що промінь від джерела світла проходить послідовно
через систему двох призм – нерухомого поляризатора та аналізатора, що
обертається. Кут обертання аналізатора визначають у градусах за допомогою
спеціального пристрою. У ході вимірювань спочатку знаходять положення
мінімальної освітленості за відсутності оптично активної речовини, а потім з
аналізованою оптично активною речовиною, що поміщена в спеціальну
поляриметричну трубку. Існують поляриметри ‒ сахариметри, у яких на
шкалі замість кута обертання показана концентрація сахарози.
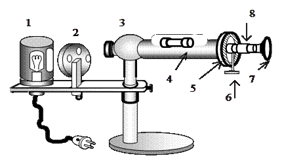
Джерелом світла в
поляриметрі є лампа розжарювання 1, світло від якої потрапляє на барабан 2,
що містить різні світлофільтри: червоний, оранжевий, зелений, синій. Після
проходження через світлофільтр світло потрапляє на вхідну головку приладу 3,
де розміщені поляризатор, конденсатор і кварцова пластинка. Далі світло
проходить через сполучну трубу 4, в яку поміщають досліджувану речовину. На
виході труби розміщений пристрій аналізатора, який складається з нерухомого
лімба 5 з градусною шкалою від 00 до 3600, двох
діаметрально розташованих ноніусів, що обертаються. Ноніуси обертаються за
допомогою фрикціона 6 та зорової труби з окуляром 7. На зоровій трубі є муфта
8, за допомогою якої встановлюється різкість бачення потрійного поля зору. Застосовують також
поляриметри інших типів, у яких аналізатор закріплений нерухомо. У таких
поляриметрах відлік кута обертання площини поляризації світла може бути
проведений точніше, ніж за поворотом аналізатора. У разі застосування таких
поляриметрів для освітлення може бути застосоване біле світло без
світлофільтрів. Поляриметричний
метод аналізу широко застосовують у харчовій
промисловості для виробництва цукру, жирів, олій. Обмеження цього методу
полягає в тому, що аналізу підлягають лише оптично активні речовини.
Контрольні питання 2. Що таке оптична активність? 3. Як розраховують величину питомого обертання речовини? 4. Поясніть принцип роботи поляриметра. 5. Як застосовують поляриметрію в контролі харчових виробництв? 4.2.3. Фотоелектроколориметрія 4.2.3.1. Закон Бугера-Ламберта-Бера
Теоретичною
основою фотометричного аналізу є закон світлопоглинання
(закон
Бугера – Ламберта – Бера).
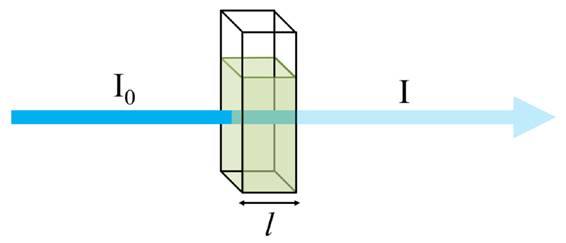
У разі проходження
світлового потоку з інтенсивністю світла I0
через розчин з концентрацією розчиненої речовини С і товщиною шару l
інтенсивність випромінювання змінюється до
I (рис. 4.6).
Рис. 4.6. Проходження світлового потоку
через розчин Математично це
виражається рівнянням:
або в
логарифмічній формі:
де Величина Залежність між
оптичною густиною поглинальної речовини та її концентрацією в розчині має
прямолінійний характер. Це чітко дотримується тільки для монохроматичних
потоків випромінювань. Величина молярного
коефіцієнта світлопоглинання залежить від: ► природи
речовини; ► довжини
хвилі монохроматичного світла; ►
температури; ► природи
розчинника.
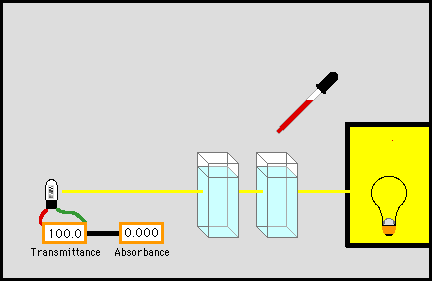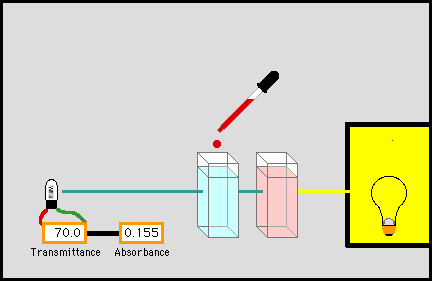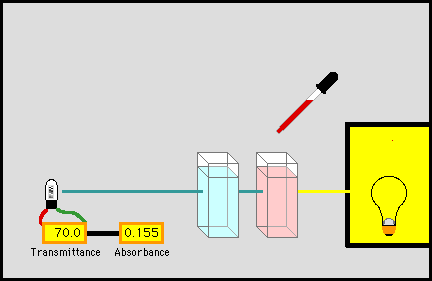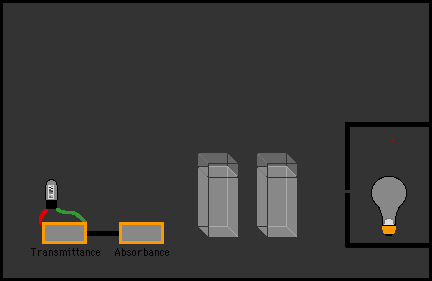
4.2.3.2. Фотоелектроколориметр У фотоколориметрії застосовують вимірювання поглинання або
пропускання світла забарвленими речовинами з використанням замість
монохроматичного випромінювання світла з вузьким інтервалом довжини хвиль.
Для цих досліджень використовують прилади – фотоелектроколориметри.
У фотоколориметрах світлова енергія перетворюється на електричну за допомогою
фотоелементів. Залежно від
кількості фотоелементів, які використовують у вимірюваннях, фотоколориметри
поділяють на однопроменеві та двопроменеві. Частіше
використовують двопроменеві фотоелектроколориметри,
оскільки вони мають вищу точність (рис. 4.7). В основу такого приладу
покладено принцип зрівнювання інтенсивності двох світлових пучків за
допомогою змінної щілистої діафрагми (принцип оптичної компенсації).
Фотоколориметр з
двома фотоелементами складається з джерела випромінювання 1, світлофільтра 2,
лінзи 3, дзеркал 4, кювет 5, діафрагм 6, фотоелементів 7, посилювача 8,
приладу для реєстрації. Існують різні види
сучасних фотоколориметрів з одним фотоелементом:
4.2.3.3. Методи визначення концентрації
розчинів за допомогою фотоелектро-колориметрії Для визначення концентрації досліджуваної речовини застосовують низку методів. Метод порівняння оптичної густини стандартного і
досліджуваного розчину. Для визначення концентрації досліджуваної речовини готують стандартний
розчин цієї речовини, у якому концентрація наближається до розчину
визначуваної речовини. Визначають оптичну густину цього розчину за певної
довжини хвилі, потім оптичну густину досліджуваного розчину за тієї самої
довжини хвилі й товщини шару. Невідому концентрацію знаходять за формулою:
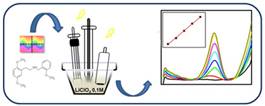
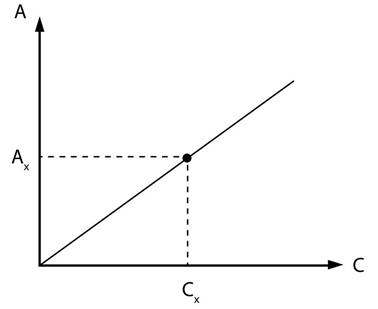
де Метод калібрувального графіка. Для визначення концентрації речовини за допомогою цього
методу готують серію із 6-8 стандартних розчинів різної концентрації.
Вимірюють оптичну густину стандартних розчинів і будують графік залежності
оптичної густини від концентрації (рис. 4.8).
Відмітивши на
графіку оптичну густину розчину з невідомою концентрацію, визначають її. Метод добавок. Метод добавок базується на порівнянні оптичної густини
досліджуваного розчину з оптичною густиною цього самого розчину з додаванням
відомої кількості визначуваної речовини. Якщо
Існують й інші
методи, що їх використовують для визначення концентрації розчинів у фотоелектроколориметрії. У лабораторіях
застосовують фотоелектроколориметри: КФК-2,
КФК-2МП, КФК-3-01, АР-101 та інші.
Контрольні питання 1. Що таке фотоелектроколориметрія? 2. Сформулюйте закон Бугера-Ламберта-Бера. 4. Поясніть принцип роботи фотоелектроколориметра. 5. Які існують фотоколориметричні методи
визначення концентрації?
4.2.4.1. Теоретичні основи
спектрофотометрії
Однією із задач
спектрофотометричного методу є кількісне визначення величин, які
характеризують поглинання цією речовиною монохроматичного випромінювання
різних довжин хвиль. Ці величини можуть бути використані як для кількісної
характеристики речовини, так і для кількісного її визначення в розчині чи в
суміші з іншими речовинами. Через поділ електромагнітного спектра за довжиною
хвилі на певні ділянки можна говорити про спектрофотометрію в інфрачервоній,
видимій і ультрафіолетовій ділянці. В ультрафіолетовій і видимій ділянці
проявляються електронні спектри молекул, в інфрачервоній ділянці – коливальні
спектри. Спектрофотометрія у видимій ділянці і УФ-ділянках дозволяє оцінювати ступінь чистоти
речовини, ідентифікувати за спектром різні сполуки, визначити константи
дисоціації кислот і основ, досліджувати процеси комплексоутворення. Інфрачервоні (ІЧ)
спектри дають характеристику речовин. Наявність в ІЧ-спектрах тих чи інших смуг
поглинання дозволяє розшифровувати структуру речовини. УФ-спектрофотометричне
вимірювання виконують у розчинах. Як розчинники використовують очищену воду,
кислоти, луги, спирти (метанол, етанол), деякі інші органічні розчинники.
Розчинник не має поглинати в тій чи іншій ділянці спектра, що й аналізована
речовина. Характер спектра (структура й положення смуг поглинання) може
змінюватися в різних розчинниках, а також за зміни рН
середовища. Вивчення спектрів
поглинання хімічних речовин з різною структурою дало можливість установити,
що основними чинниками, які зумовлюють поглинання світла, є наявність так
званих хромофорів, тобто ненасиченість (подвійні чи
потрійні зв’язки), наявність карбонільної, карбоксильної,
амідної, азо-, нітрозо-, нітро- та інших функціональних груп. Кожна функціональна
група характеризується поглинанням у певній ділянці спектра. Але є певні
чинники (наявність декількох хромофорних груп, вплив розчинника та ін.), які
призводять до зміщення смуг поглинання в бік більших довжин хвиль (батохромне
зміщення) або в бік коротких довжин хвиль (гіпсохромне зміщення). Крім
зміщення, може спостерігатися ефект збільшення (гіперхромний)
чи зменшення (гіпохромний) інтенсивності
поглинання. У зв’язку з цим
для ідентифікації речовини за її УФ-спектром застосовують метод порівняння із
спектром відомої речовини, одержаним за тих самих умов. Характеристикою
спектра поглинання речовини є положення максимумів (мінімумів) поглинання, а
також інтенсивність поглинання, що характеризується величиною густини чи
питомого показника поглинання за конкретної довжини хвилі. Метод
ІЧ-спектроскопії дає можливість одержати найбільш повну інформацію про будову
та склад досліджуваної речовини, яка дозволяє ідентифікувати дуже близькі за
структурою сполуки. Метод інфрачервоної спектроскопії прийнятий для
ідентифікації органічних речовин з поліфункціональними
групами шляхом порівняння із спектрами стандартних зразків, які зняті в
однакових умовах. Спектрофотометричні
методи дають можливість кількісно визначати речовину в широкому інтервалі
довжини хвиль – від 185 до 1100 нм, здійснювати аналіз багатокомпонентних
систем. На відміну від
фотометрії, у спектрофотометрах монохроматизація світла забезпечується не
світлофільтрами, а монохроматором, що дозволяє безперервно змінювати довжину
хвилі. Монохроматорами є дифракційні ґрати або призми, які забезпечують більш
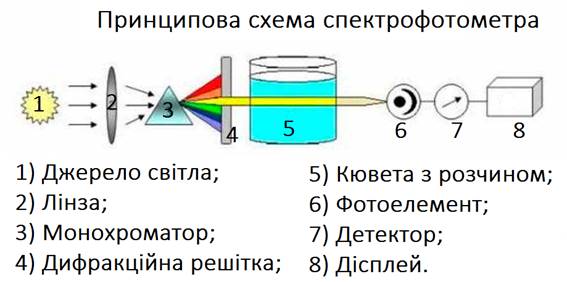
високу точність спектрофотометричних досліджень. Спектрофотометр — це здебільшого сукупність монохроматора, який
призначений для виділення необхідної вузької спектральної ділянки світлового
пучка, та вимірювального фотометра зі звичайною або автоматичною реєстрацією.
Спектрофотометрами
вимірюють коефіцієнти пропускання (або оптичної густини) твердих тіл, шарів
рідини на дуже вузькій ділянці спектра. Деякі сучасні спектрофотометри є
автоматичними приладами, які записують інформацію про спектральні коефіцієнти
пропускання зразка, який досліджують, у формі графіка або числової таблиці. Спектральна ширина
смуги, яка виділяється монохроматором, встановлює по суті межу дозволу.
Останнє визначає роздільну здатність спектрофотометра λ/Δλ. Удосконалення цих приладів і спрямовано на
збільшення їхньої роздільної здатності й відповідно зменшення спектральної
ширини діючого випромінювання, що пов’язано зі зменшенням потужності
випромінювання й електричного струму, який виникає під дією випромінювання в
зовнішньому колі приймача. Тому до складу кожного сучасного спектрофотометра
входить пристрій, призначений для посилення, вимірювання, а інколи й
реєстрації фотоструму. Слід зауважити, що
теоретично не можна отримати чітко монохроматичне випромінювання, яке завжди
має деякий кінцевий спектральний інтервал, що визначається явищами дифракції
і аберації. У
світловимірювальній практиці використовують такі прилади: 1) спектрофотометр
СФ-10, який вимірює світлотехнічні характеристики в діапазоні 400-750 нм; 2) спектрофотометр
СФ-20, який вимірює спектральну характеристику зразків на ділянці спектра від
186 до 2500 нм, тобто охоплюючи ділянку ІЧ-випромінювання; 3) спектрофотометр
СФ-46, який вимірює коефіцієнти пропускання прозорих і плоскопаралельних
зразків у спектральній ділянці 190-1100 нм. Розглянемо
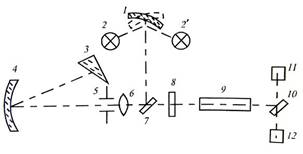
докладніше схему й принцип дії дуже поширеного сьогодні компактного
спектрофотометра СФ-46, оптичну схему якого наведено на рис. 4.9. Джерелом світла є
вольфрамова лампа розжарювання, яка освітлює конденсорне дзеркало 1. З
останнього промені світла падають на плоске дзеркало 7, у свою чергу,
спрямовуючи світловий потік на вхідну щілину 5, яка розташована у фокусі
дзеркального сферичного об’єктива 4. Відбитий від сферичного дзеркала 4
паралельний пучок променів падає на поверхню кварцової призми 3, заломлюється
в ній і падає майже по нормалі на її другу грань, яка покрита зовні шаром
алюмінію. Після відбиття пучок променів удруге проходить крізь призму, вдруге
заломлюється на її першій грані. Як результат такого проходження різні
спектральні складові випромінювання виходять із призми в різних напрямках.
Повертаючи призму 3 у межах деякого кута навколо осі, яка перпендикулярна
граням призми, можна спрямувати на сферичне дзеркало 4 паралельні пучки тієї
чи іншої частини спектра. Відбиті від дзеркала 4 пучки променів крізь вихідну
щілину й лінзу, які розташовані вище вхідної щілини 5 (на рисунку не
показано), потрапляють на дзеркало 10. Поворотне дзеркало
10 може спрямовувати пучок променів на один з приймачів випромінювання 11 або
12. Приймачем 11 є сурм’яно-цезієвий фотоелемент
Ф-17, приймачем 12 – киснево-цезієвий фотоелемент Ф-23. Фотоелемент Ф-17
застосовують під час вимірювання випромінювання в ділянці від 186 до 650 нм,
Ф-23 – в ділянці від 600 до 1100 нм. У цьому випадку вимірюємо значення
фотоструму, яке відповідає значенню 100 % світлового потоку, що вільно
проходить.
Під час виконання
вимірювання коефіцієнтів пропускання прозорих тіл або суміші на шляху між
вхідною щілиною приладу й дзеркалом 10 встановлюють скляний світлофільтр 8 і
кювету 9 зі сумішшю. У кюветі можуть розташовуватися водночас три дослідних
зразки з різною оптичною густиною, вимірювання яких можна робити почергово.
Світлофільтр 8 застосовують для затримання розсіяного світла сторонніх довжин
хвиль. Наприклад, під час вимірювання в ділянці 320-380 нм за світлофільтр
застосовують скло УФС-2, яке пропускає УФ-промені й затримує промені видимої
частини спектра. Спектральний
інтервал Δλ, який виділяється приладом,
різний у різних частинах спектра й залежить від ширини вхідних і вихідних
щілин. Електрична схема
спектрофотометра СФ-46 забезпечує живлення джерел світла стабільною напругою,
посилюванням фотоструму, який виникає у фотоприймачі під дією пропущеного
випромінювання. Крім описаних приладів, набули поширення й сучасні
спектрофотометри, які можна використовувати разом з комп’ютерною технікою. Кількісний аналіз однокомпонентних систем виконують тими самими методами, що й у фотоелектроколориметрії: ► методом
порівняння оптичної густини стандартного й досліджуваного розчинів; ► методом
калібрувального (градуювального) графіка та іншими. Спектрофотометричні
вимірювання можна використовувати для фіксації точки еквівалентності в
титруванні. Кінцева точка прямого фотометричного титрування з’являється
внаслідок зміни концентрації реагенту й продукту реакції. Крива
фотометричного титрування є графіком залежності оптичної густини від об’єму титранту. За правильно вибраних умов, крива складається з
двох прямолінійних ділянок з різним нахилом: одна з них відповідає початку
титрування, інша – продовженню за точкою еквівалентності. Поблизу точки
еквівалентності спостерігається помітний перегин. Кінцевою точкою вважають
точку перетину прямолінійних відрізків після екстраполяції.
Контрольні питання 2. У якій ділянці спектра дозволяє проводити дослідження
спектрофотометрія? 3. Поясніть принципову схему спектрофотометра. 4. Якими методами користуються у спектрофотометрії? 4.3. Електрохімічні методи аналізу 4.3.1.1. Теоретичні основи кондуктометрії
Розчини
електролітів мають електричний опір R,
який на ділянці завдовжки l із
перерізом S визначають
співвідношенням:
де Величина L, обернена електричному опору,
називається електричною провідністю:
Одиницею вимірювання електричної провідності є сіменс (См): 1 См = 1 Ом-1. Величина, обернена питомому електричному опору, називається питомою електричною провідністю (χ):
Одиницею вимірю
цієї величини є См·см-1. Питома електрична
провідність сильного електроліту КА, який дисоціює на йони
К+ і А-, дорівнює
де
Рівняння для
питомої електричної провідності слабкого електроліту має вигляд:
де
Питома електрична
провідність електроліту зростає зі збільшенням концентрації його розчину до
деякої межі, проходить через максимум і починає зменшуватися. Це пояснюється
тим, що зі зростанням концентрації збільшується міжйонна
взаємодія та зменшується ступінь дисоціації слабких електролітів. Тому в кондуктометричних дослідженнях використовують лише ту
концентраційну ділянку, де спостерігається лінійна залежність між
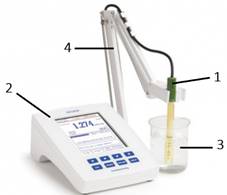
концентрацією й електричною провідністю розчину. Кондуктометричні вимірювання виконують за допомогою кондуктометрів. Сучасні
кондуктометри (рис. 4.10) призначені для автоматичного вимірювання питомої
електричної провідності розчинів.
Кондуктометри
мають двоконтактні датчики з платиновими
електродами 1, результати вимірювань яких передаються на основний блок 2 з
дисплеєм. Досліджуваний розчин міститься в скляному стакані 3. Датчик з
електродами й стакан закріплюють в штативі 4. Кондуктометр живиться від блоку
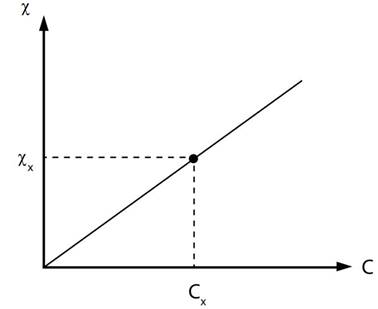
живлення. Пряма
кондуктометрія базується на безпосередньому визначенні концентрації розчину
електроліту за виміряною величиною питомої електричної провідності χ .
Для цього готують серію розчинів цього електроліту з різною концентрацією. На
підставі вимірювань питомої електричної провідності стандартних розчинів
будують калібрувальний графік. Електричну провідність електроліту з невідомою
концентрацією вимірюють у тих самих умовах, що й під час побудови графіка. За
допомогою калібрувального графіка визначають концентрацію досліджуваного
розчину (рис. 4.11).
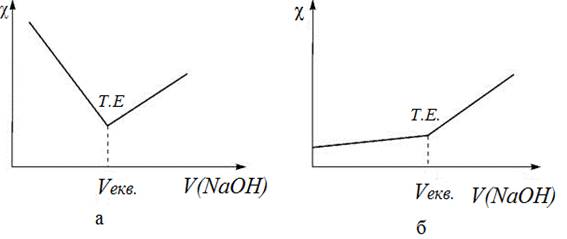
4.3.1.4. Кондуктометричне титрування Кондуктометричне титрування базується на визначенні точки еквівалентності за різкою
зміною питомої електричної провідності розчину. Важливою умовою цього
методу є те, що електрична провідність
розчину, який титрують, має значно відрізнятися від електричної провідності титранту або продуктів реакції. Кондуктометричне титрування використовують для кислотно-основного титрування, осаджувального, комплексонометричного
титрування. Його часто застосовують у тих випадках, коли розчин має
інтенсивне забарвлення й каламутний та індикаторний метод фіксації точки
еквівалентності неможливі. Кондуктометрично
визначають концентрацію органічних кислот у фруктових соках, безалкогольних
напоях та в різних видах кулінарної продукції. У разі
використання кондуктометричного титрування титрант додають невеликими порціями, і після кожного
додавання вимірюють питому електричну провідність χ. На підставі
одержаних даних будують криву титрування в координатах χ – V титранту.
Точку еквівалентності визначають у місці перетину прямих ліній, що відповідають
зміні питомої електричної провідності розчину до і після точки
еквівалентності. Розглянемо два
види кривих титрування.
У разі титрування
сильної кислоти сильною основою (рис. 4.12, а) у початковому розчині питома електрична провідність має велике
значення, оскільки в такому розчині є катіони Н+, які мають
високе значення еквівалентної електричної провідності катіона. Під час
титрування концентрація Н+
зменшується внаслідок нейтралізації:
Мінімальне
значення χ буде в точці еквівалентності. Додавання надлишку лугу-титранту призводить до зростання електричної провідності
унаслідок появи в розчині надлишку високопровідних
аніонів OH-. У разі титрування
лугами слабких кислот (рис. 4.11б), наприклад ацетатної, електрична
провідність розчину на початку титрування буде низькою через невисокий
ступінь дисоціації кислоти. З додаванням титранту
питома електрична провідність розчину повільно зростатиме внаслідок утворення
в розчині сильного електроліту – натрій ацетату. Вплив Н+ йонів на електричну
провідність розчину до точки еквівалентності буде невеликою внаслідок
утворення ацетатної буферної суміші. Після точки еквівалентності електрична
провідність розчину різко зросте внаслідок появи в розчині високопровідних OH-
йонів. Кондуктометричне титрування має певні переваги перед методом прямої кондуктометрії:
висока селективність, можливість диференційного титрування сумішей кислот або
основ, можливість титрування каламутних і забарвлених розчинів.
Контрольні питання 2. Що таке електропровідність? 3. Що таке питома електропровідність? 4. Що таке еквівалентна електропровідність? 5. Поясніть принцип прямої кондуктометрії. 6. Поясніть принцип кондуктометричного
титрування. 4.3.2.1. Теоретичні основи потенціометрії
Потенціометричні
методи дозволяють визначити концентрацію певних йонів
у розчині. Взаємозв’язок між концентрацією йонів
(активністю) і рівноважним значенням електродного потенціалу описується
рівнянням Нернста:
де Для температури
298 К і з переходом від натурального логарифма до десяткового формула набуває
вигляду:
У разі занурення
інертного платинового електрода в розчин, що містить речовину в різних формах
окиснення, металічний електрод виконує роль передавача електронів і з ним не
відбувається змін. Стрибок потенціалу, що виникає на межі електрод – розчин
називають окисно-відновним потенціалом; значення його згідно з рівнянням Нернста:
де 4.3.2.2. Електроди потенціометрії У
потенціометричному аналізі використовують електроди двох типів: - електроди, на
поверхні яких відбуваються реакції з обміном електронів. Їх називають
окисно-відновними. Такі електроди виготовляють з платини. Зараз
використовують електроди зі спеціального скла – мембранний окисно-відновний
електрод; - електроди, на
поверхні яких відбуваються реакції обміну йонів. Їх
називають йонообмінними або йонселективними
електродами. Їх також називають мембранними. Йонселективні електроди поділяють залежно від
типу мембран на такі типи: ► електроди
з твердими мембранами; ► електроди
із скляними мембранами; ► електроди
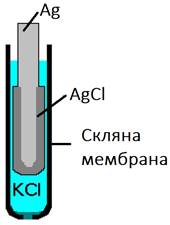
з рідинними мембранами. Розглянемо будову скляного хлорсрібного електрода (рис. 4.13).
Скляний електрод
виготовляють зі спеціального скла, до складу якого входять оксиди алюмінію,
натрію, калію, бору та інших металів. Мембрана такого електрода є
тонкостінною кулькою (0,1 мм) діаметром 5-8 мм. Усередині такого електрода є
срібна дротинка, вкрита шаром арґентум хлориду, яка розташована в насиченому
розчині калій хлориду. Метод прямої
потенціометрії базується на визначенні концентрації аналізованого йона за виміряною ЕРС електрохімічного кола, що містить
відповідний йонселективний електрод. Цей метод широко
застосовують для визначення рН розчинів. Для
вимірювання рН у досліджуваний розчин занурюють
систему з двох електродів: скляного та електрода порівняння, наприклад хлорсрібного. Потенціал електродів вимірюють за допомогою
рН-метрів різних марок. Для визначення
концентрації аналізованого йона користуються
методом калібрувального графіка. За цим методом на підставі вимірювань ЕРС
для стандартних розчинів будують графік у координатах: ЕРС – lg С. Такий графік має лінійний
вигляд. Потім вимірюють ЕРС кола з аналізованим розчином і за графіком
визначають концентрацію.
Рис.4. Х. рН -
метри 4.3.2.4. Потенціометричне титрування Метод
потенціометричного титрування базується на вимірюванні точки еквівалентності
за різкою зміною ЕРС в електрохімічному колі, що містить електрод порівняння.
Індикаторний електрод обов’язково має бути селективним до визначуваних йонів. У методі
потенціометричного титрування використовують усі види титрувань:
кислотно-основне, осаджувальне, окисно-відновне, комплексонометричне. Точку
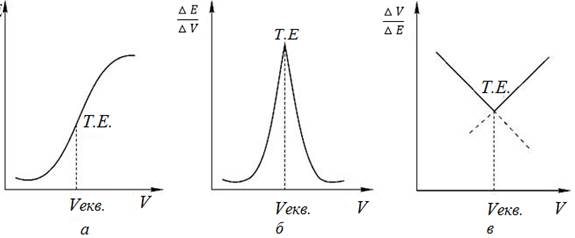
еквівалентності визначають графічним методом на кривій титрування.
Розрізняють такі криві титрування: інтегральну, диференціальну та криву Грана
(рис. 4.14).
Інтегральна крива
титрування будується в координатах Диференціальна
крива титрування будується в координатах: Крива титрування
Грана будується в координатах: Точка еквівалентності
відповідає об’єму титранта на перетині двох прямих
ліній. Цією кривою зручно користуватися для визначення точки еквівалентності
під час титрування розбавлених розчинів. Потенціометричне
титрування використовують в аналітичному контролі харчових виробництв, для
визначення титрованої кислотності харчових продуктів (хліб, напої, молоко,
молочні продукти). Метод потенціометричного титрування вирізняється точністю,
можливістю аналізувати розбавлені розчини, можливістю виконувати титрування
каламутних та забарвлених розчинів. Також цей метод дозволяє автоматизувати
процес.
Контрольні питання 2. Які електроди використовують в потенціометрії? 3. Яку будову має скляний електрод? 4. Що таке пряма потенціометрія? 5. Що таке потенціометричне титрування? 4.4. Хроматографічні методи аналізу 4.4.1. Сутність та класифікація
хроматографічних методів аналізу 4.4.1.1. Теоретичні основи
хроматографічного аналізу Хроматографічний
метод відкрив у 1903 р. М. Цвєт, який вперше
застосував його для розділення рослинних пігментів. Першим рослинним
пігментом був хлорофіл.
Хроматографічні
методи ґрунтуються на сорбційних процесах.
Розрізняють
поглинання поверхневим шаром твердого тіла або рідини (адсорбцію) і поглинання
всім об’ємом сорбенту (абсорбцію). Різна здатність до
поглинання певним сорбентом різних компонентів сорбату
спричинює різну швидкість розповсюдження компонентів у поглинальній речовині
і, як наслідок, розділення суміші.
Конструкційно розділення здійснюється в обмеженому об’ємі (дуже часто це циліндр або
так звана колонка), заповненому пористою гранульованою твердою речовиною (нерухома
фаза), через яку рухається (рухома фаза) носій (рідина або
газ) з досліджуваною сумішшю. Нерухомою фазою може бути також плівка рідини
на твердому тілі. На вхід (згори) вертикально розташованої колонки подається
рухома фаза з сумішшю, а на виході (внизу) спостерігаємо послідовну появу
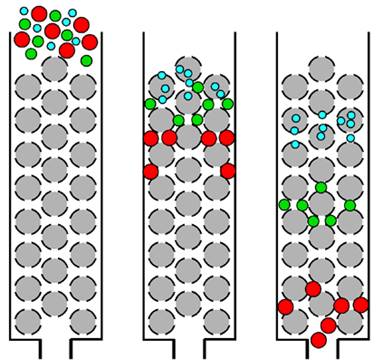
різних компонентів суміші (рис. 4.15). Оскільки процес переміщення рухомої
фази під дією сил тяжіння повільний, то для його прискорення створюють
додаткові тиски (іноді до сотень атмосфер). На виході хроматографічної
колонки компоненти детектують (визначають за
допомогою детекторів різної природи, наприклад спектральних приладів).
Наприклад, маємо
колонку (див. рис. 4.16), заповнену гранульованою твердою речовиною, через
яку протікає потік рідини з розчиненими у ньому речовинами А (компонент 1) та В (компонент 2). Кожна молекула
речовин А та В певний час перебуває в потоці рухомої фази, а решту часу
внаслідок адсорбції утримується на поверхні нерухомої фази. Відношення
концентрації речовини А чи В у нерухомій фазі до концентрації
тієї самої речовини в рухомій фазі є константою
(коефіцієнтом) розподілу для цієї речовини. Можливість розділення
речовин А та В зумовлена тим, що одна з них перебуває більше часу в рухомій
фазі й швидше досягне кінця колонки. Швидкість руху зони певної речовини
через колонку, яка визначає цей час, є обернено пропорційною коефіцієнтові
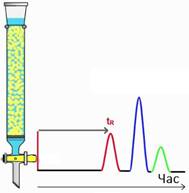
розподілу. Якщо на виході колонки реєструвати зміну в часі чи об’ємі рухомої
фази будь-яку її властивість, то на стрічці реєстратора отримаємо вихідну
хроматографічну криву – хроматограму (рис. 4.17).
Сорбційна здатність
нерухомої фази за відношенням до речовин, які розділяємо, характеризується
часом утримання tR. Час утримання – це відстань на хроматограмі
від моменту введення проби у шар сорбенту до моменту появи на виході із цього
шару речовини максимальної концентрації у потоці рухомої фази. Об’єм рухомої
фази, який пройшов при цьому через шар сорбенту, є об’ємом утримання VR.
Цей об’єм і час утримання пов’язані співвідношенням:
де Час утримування –
якісна характеристика речовини. Висота вихідної кривої (піка) h є довжиною перпендикуляра від максимуму піка на нульову
лінію. Нульова лінія відповідає сигналу детектора за виходу рухомої фази без
проби. Це – кількісна характеристика речовини. Ширина піка m – відрізок на
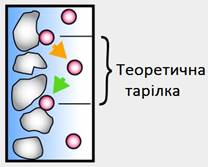
нульовій лінії, утворений дотичними до кривої в точках її перегинів. Під час хроматографування утворюються зони розділених речовин. Ці
зони під час руху в колонці розмиваються. Що більша розмитість зон, то важче
їх розділити. Мірою розмитості є так звана висота еквівалентної теоретичної
тарілки ВЕТТ.
Що більше подібних
станів рівноваги, то кращим є розділення. 4.4.1.2. Класифікація хроматографічних
методів аналізу Хроматографічні
методи розрізняють за такими ознаками: ► за
середовищем, у якому виконують розділення: газова, газорідинна та рідинна хроматографія; ► за
механізмом розділення: молекулярна, ситова, йонообмінна,
осадова, розподільна хроматографія; ► за формою
проведення процесу: колонкова, паперова, тонкошарова,
капілярна хроматографія. Залежно від
способу переміщення досліджуваної суміші вздовж шару сорбенту
хроматографічний процес може бути фронтальним, проявниковим
та витиснювальним.
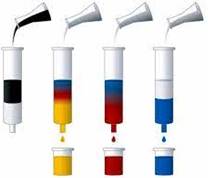
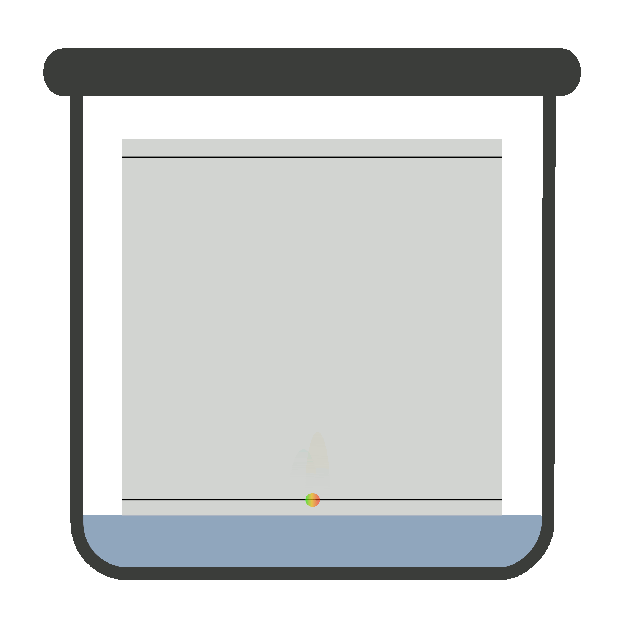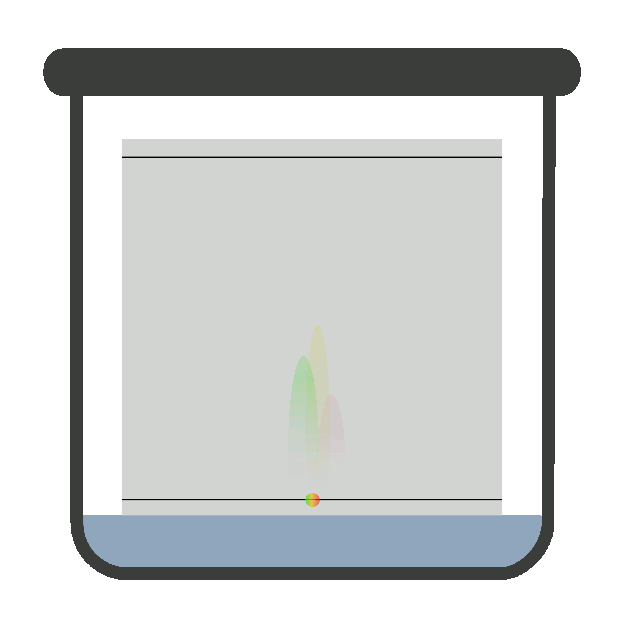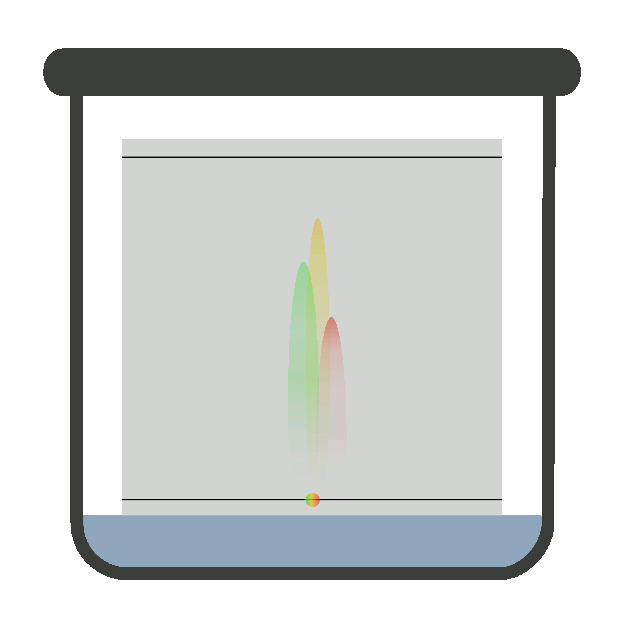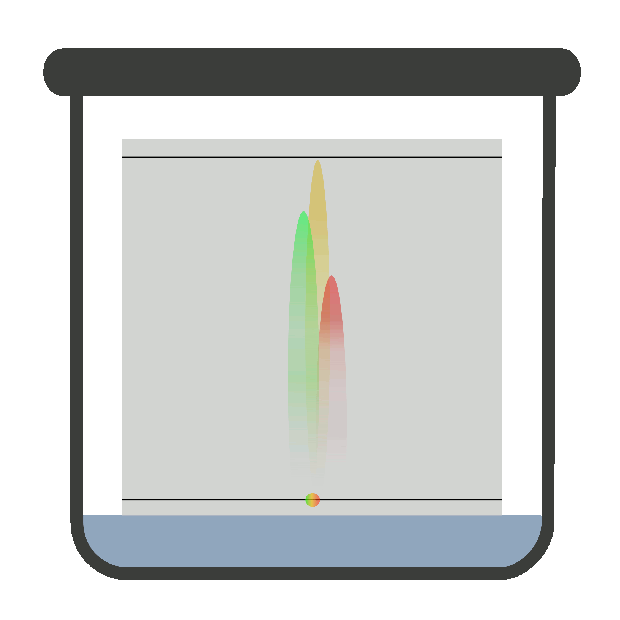
Контрольні питання 3. Що таке коефіцієнт розподілу? 5. Що таке висота вихідної кривої? 7. Як класифікують хроматографічні методи аналізу? 4.4.2. Основні методи хроматографічного аналізу 4.4.2.1. Паперова тонкошарова хроматографія У дослідженнях
широко застосовують паперову й тонкошарову хроматографії, які відрізняються
від інших хроматографічних методів простотою та зручністю виконання
експерименту. У поєднанні з мікрокількістю
досліджуваних речовин, необхідних для аналізу, це забезпечило її значне
поширення в хімічному аналізі. Паперову хроматографію (ПХ) поділяють на розподільну, адсорбційну та йонообмінну.
У ПХ як нерухому фазу використовують хроматографічний папір або речовини,
заздалегідь нанесені на його волокна. Механізм хроматографії на папері може
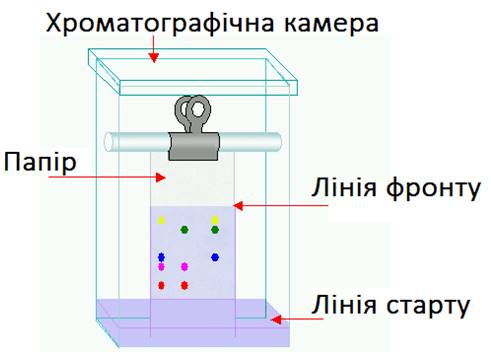
бути розподільним або адсорбційним. Для одержання хроматограми розчини чистих речовин («свідків») і суміші,
яку необхідно розділити, наносять на хроматографічний папір, нижній кінець
якого занурюють у відповідну систему розчинників. Через деякий час суміш
розділяється на зони окремих компонентів. Для виявлення зон хроматограму розглядають у світлі УФ-випромінювання за
певної довжини хвилі і позначають олівцем контури плям. Якщо компоненти дають
кольорові реакції, то хроматограму проявляють,
занурюючи її в розчин реагенту або обприскуючи з пульверизатора. Характеристикою
компонентів є величина Rf – відношення відстані від стартової
лінії хроматограми до центра плями цієї речовини l до відстані, яку пройшов фронт
розчинника: Rf
= l : L. Типову хроматограму
наведено на рис. 4.17. Величину Rf
використовують для ідентифікації речовин. Ідентичність визначають
одночасним хроматографуванням на одному аркуші
паперу досліджуваного й автентичного зразка тієї самої речовини. Якщо зразки
ідентичні, відповідні їм плями на хроматограмах
мають однаковий вигляд і однакові значення Rf.
Рис. 4.17. Хроматограма
розділення багатокомпонентної суміші 4.4.2.2. Газова хроматографія Для розділення,
аналізу й дослідження речовин та їх сумішей, що не розкладаються в
газоподібному стані, найбільшого застосування набула газова хроматографія.
Залежно від виду сорбенту, яким заповнюють хроматографічну колонку, її
поділяють на газоадсорбційну та газорідинну. У газовій
хроматографії як рухому фазу (газ-носій) найчастіше використовують інертні
гази. Процес розділення
й аналізу виконують за допомогою спеціальних приладів, які називають газовими хроматографами. Незважаючи на
різні технічні рішення, рівень забезпечення електронними вузлами та основні
технічні характеристики, принцип будови хроматографів однаковий. У кожному з
них є такі основні вузли: система подавання газу-носія, пристрій для уведення
досліджуваної суміші, хроматографічна колонка, аналізатор (детектор), прилад
для реєстрації та аналізу. Під час
проходження досліджуваної суміші через хроматографічну колонку її компоненти селективно (вибірково) утримуються нерухомою фазою, а
потім виходять із колонки і реєструються детектором, сигнали якого
автоматично записуються у вигляді хроматограми. 4.4.2.3. Рідинна хроматографія У методі рідинної
хроматографії рухомою фазою є рідина, нерухомою – твердий адсорбент. На
відміну від газової, рідинна хроматографія може бути використана для аналізу
речовин з молекулярною масою від кількох сотень до кількох мільйонів,
включаючи складні макромолекули нуклеїнових кислот, білків тощо.
Рис. 4.19. Рідинна хроматографія Залежно від
характеру взаємодій, що відбуваються в шарі сорбенту, рідинну хроматографію
поділяють на дві групи: молекулярну та хемосорбційну. До першої групи
відносять молекулярно-ситову (гель-фільтраційну), адсорбційно-рідинну
та рідинно-рідинну хроматографію з відносно слабкою взаємодією в системі сорбат – сорбент – розчинник. До другої групи
належать йонообмінна, комплексоутворювальна,
осадова, окисно-відновна та афінна хроматографія – із сильною взаємодією між сорбатом і сорбентом. Молекулярна рідинна хроматографія. Під час виконання аналізу цим методом використовують
колонки значно меншої довжини, ніж у газовій хроматографії. Як розчинник
застосовують легкі вуглеводні та їх похідні: гексан, бензол, толуол, метанол,
етанол, ацетатну кислоту тощо. Їх вибір визначається типом сорбенту, яким
може бути силікагель, алюміній оксид, магній оксид, сахароза, полімери. Адсорбційно-рідинний та рідинно-рідинний
методи хроматографії
тісно пов'язані між собою. Їх застосовують для розділення сумішей
нуклеотидів, вітамінів, лікарських препаратів та інших складних органічних сполук. Молекулярно-ситова (гель-фільтраційна) хроматографія ґрунтується на принципі розділення суміші речовин за
розмірами їх молекул. Процес відбувається за рахунок того, що крізь цеоліт
(молекулярне сито) або пори гелю можуть дифундувати тільки речовини, розміри
молекул яких не перевищують розміри пор адсорбенту. Внаслідок цього молекули
меншого розміру проходять більший шлях і виходять з колонки пізніше, ніж
більші молекули.
Рис. 4.20. Молекулярно-ситова
хроматографія Останнім часом
молекулярно-ситову хроматографію широко використовують для визначення
молекулярної маси білків, виділення та очищення біополімерів (білки, пептиди,
полісахариди, нуклеїнові кислоти). Тонкошарова хроматографія. У методі
тонкошарової хроматографії (ТШХ) роль носія відіграє тонкий шар
порошкоподібного сорбенту, нанесений на скляну чи металеву пластинку. Як
сорбенти застосовують силікагель, алюміній оксид, магній силікат тощо.
Рис. 4.21. Тонкошарова хроматографія Методика аналізу
загалом мало чим відрізняється від методики проведення ПХ. Водночас метод ТШХ
має низку переваг: висока швидкість процесу хроматографування,
можливість використання як нерухомої фази (носія) різних сорбентів,
застосування кислих та лужних рухомих фаз та оброблення хроматограм
за підвищених температур. ПХ і ТШХ посідають
одне з провідних місць серед методів розділення й аналізу органічних та
біоорганічних сполук. Ними можна визначати речовини
масою 10-20 мкг, тривалість розділення становить
кілька хвилин, тому їх часто застосовують як експрес-методи.
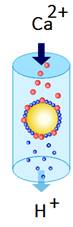
4.4.2.4. Йоннообмінна хроматографія Йонообмінна хроматографія ґрунтується на оборотності хемосорбції йонів
досліджуваного розчину йоногенними групами
сорбенту. Процес обміну йонів у системі
сорбент–розчинник відбувається в стехіометричних
співвідношеннях. Сорбенти, здатні
до йонообмінної адсорбції, називають йонітами. Залежно від
характеру йоногенних груп їх поділяють на катіоніти та
аніоніти.
Катіоніти ‒ нерозчинні високомолекулярні сполуки, що містять у своєму складі кислотні групи різної сили. За ступенем
йонізації йоногенних груп
їх поділяють на сильнокислотні – КУ-2, СДВ-3 (до їх
складу входять переважно залишки сульфатної та фосфатної кислот) та слабкокислотні – КБ-4, КБ-4П-2, що містять карбоксильні, сульфгідрильні та інші малодисоційовані
кислотні групи. Аніоніти – також нерозчинні ВМС, до складу яких входять основні групи. За ступенем
кислотності аніоніти поділяють на сильноосновні –
АВ-16, АВ-17 (йоногенні групи – амонієві основи) і слабкоосновні – АН-2Ф, ТМ (йоногенні
групи – аміногрупи та залишки інших слабких органічних основ). У водних розчинах йоніти поглинають значну кількість води (до 50 %) і
набухають. Це призводить до гідратації йоногенних
груп та їх йонізації. Катіоніти в Н-формі й аніоніти в ОН-формі можуть відповідно
обмінюватися тільки йонами Гідрогену чи
гідроксид-іонами. У сольових формах йонітів катіони
Гідрогену заміщені на катіони металу чи органічної основи, а гідроксид-іони –
на аніони кислот. Для хроматографування застосовують
як сольові йоніти, так і в Н- або ОН-формах. Оскільки йонний обмін – процес оборотний, це дає можливість
здійснювати регенерацію йонітів. Катіоніти регенерують кип'ятінням у розчині сильних
кислот, аніоніти – відповідно в розчині лугів. Здатність йонітів до йонообмінної
адсорбції характеризується активністю, яку оцінюють за об'ємною здатністю. Об'ємна здатність – це кількість
молів йонів, що зв'язується за рівноважних умов
сухим йонітом масою один грам. Йонообмінні методи застосовують як для визначення сумарного вмісту катіонів чи
аніонів у розчині, так і для аналізу розчинів чистих солей. Так, під час
пропускання крізь катіоніт Н-форми розчину солі Натрію в ньому як результат йонного обміну з'являється еквівалентна кількість йонів Гідрогену, які можна визначити титриметричним
методом. Велике практичне
значення має процес демінералізації води та різних
сольових розчинів, в основі якого лежить йонообмінна
адсорбція. Воду або сольовий розчин пропускають почергово крізь катіоніт Н-форми
та аніоніт ОН-форми. Як результат йонного обміну в розчині утворюється еквівалентна
кількість йонів Гідрогену та гідроксид-іонів, які
взаємодіють між собою з утворенням води. Це призводить до зміщення рівноваги йонного обміну й повного вилучення солей з розчину. Таку
воду називають демінералізованою, або знесоленою.
Йонообмінну хроматографію широко застосовують для розділення неорганічних і органічних
речовин. Цим методом можна відокремити дуже близькі за хімічними
властивостями рідкісноземельні елементи, амінокислоти, ферменти та інші
сполуки.
Контрольні питання 1. Поясніть принципи паперової та тонкошарової хроматографії. 2. Поясніть принцип газової хроматографії. 3. Що є рухомою і нерухомою фазою в рідинній хроматографії? 4. Чим відрізняється молекулярна рідинна хроматографія від газової? 5. Поясніть принцип молекулярно-ситової хроматографії. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||